
Molecular Modeling & Drug Discovery
6. Protein Structure Prediction:
Protein Structure Prediction
Prediction of protein structure involves three main strategies:
-
Homology and Comparative Modeling
-
Recognizing Folds Computationally
-
Structure Prediction from Ab lnitio
Modeling through homology
Use a protein with a known structure as a structural template to create the modeling target based on the protein's similarity in sequence.
Therefore, "homology modeling" is more appropriately referred to as "comparative modeling" since template and target proteins need not be connected in terms of evolution.
The most crucial phase of comparative modeling is the creation of topologically correct sequence alignments.
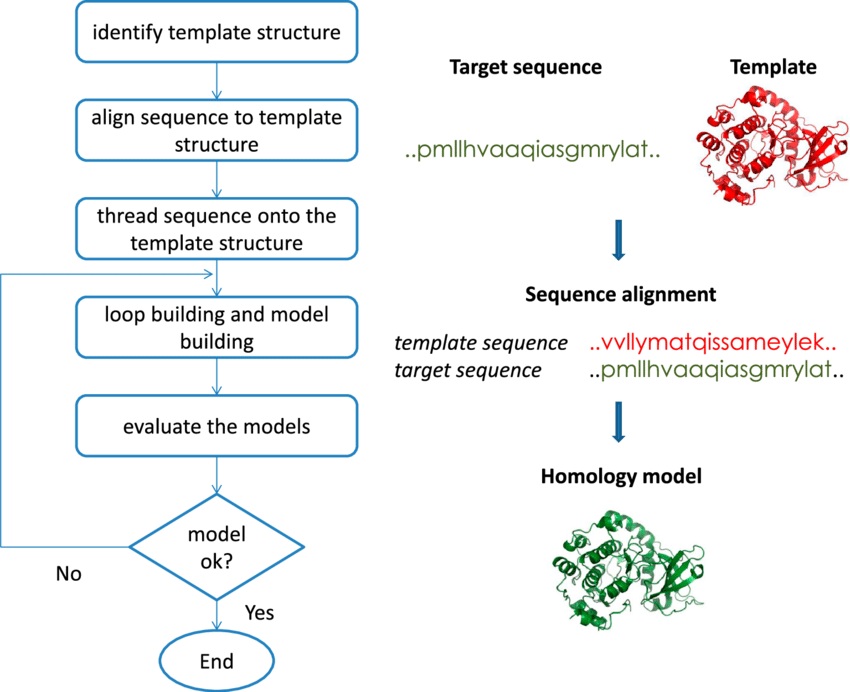
Figure 1. One of the computer methods for predicting the 3D structure of a protein from its amino acid sequence is homology modeling. It is regarded as the computational structure prediction method with the highest level of accuracy. It comprises of a number of simple and easy to follow steps.
The main factor affecting how accurate a homology model is alignment.
No calculations for computational refinement will locate mismatched residues in their proper spatial locations.
Wrong structures were produced by misaligned sequences.
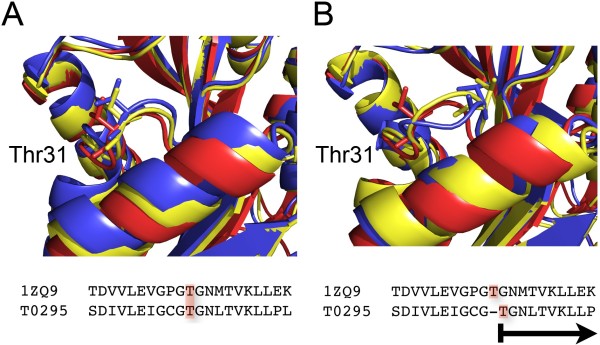
Figure 2. The crystal structure of human dimethyladenosine transferase (PDB 1ZQ9) is used as a model for the target protein T0295 from CASP7.
Analysis of figure 2:
In (A), we display how the best (yellow) and poorest (blue) models produced by MODELLER 9v5 for T0295 structurally correspond with the template 1ZQ9.
3D-Figure 1. PDB structure of 1ZQ9 is of human Dimethyladenosine transferase with Two chains A,B (CLICK me for more info!!).
For instance, Thr31 superposes in all three structures quite successfully.
In Figure (B), we demonstrate the impact of a mistake in the sequence alignment used as the modelling process' input. We created new models for T0295 after shifting the alignment by one residue at the level of Thr31.
Due to a single mistake in the sequence alignment, Thr31 is shifting by 3.4 Å (Armstrong).
Superposition compared with Alignment
In three dimensions, two proteins' coordinates can be superimposed.
The residues that are in equivalent locations in space are identified from a superposition to create a structure-based sequence alignment.
A sequence alignment that is based on statistics is more likely to produce a different alignment outcome.
Superposition of '1GTR' and '2TS1' leds us to determine similar structure and function for both.
3D-Figure 2. PDB structure of 2TS1 is of RNA Synthetase/TYROSYL-T with chain A and sequence length of 419 (CLICK me for more info!!).
3D-Figure 3. PDB structure of 1GTR is of Glutaminyl-tRNA Synthetase Anticodon Loop Recognition with chains B [auth A] of sequence length 553 (CLICK me for more info!!).
Conducting alignment on sequence :
.png)
.png)
Figure 3. Depiction of pairwise alignment of structures(left) and residues(right) for PDB entries 1GTR and 2TS1.
| Colors | Entry ID | Chain ID | Description | Organism | Sequence Length | Modeled Residues |
|---|---|---|---|---|---|---|
| 1GTR | B [auth A] | GLUTAMINYL-tRNA SYNTHETASE | Escherichia coli | 553 | 529 | |
| 2TS1 | A | TYROSYL-TRNA SYNTHETASE | Geobacillus stearothermophilus | 419 | 317 |
| RMSD | TM-score | Sequence Identity | Equivalent Residues | Reference Coverage | Target Coverage |
|---|---|---|---|---|---|
| 4.34 | 0.33 | 9% | 212 | 40% | 67% |
Despite the statistically ideal sequence
alignment having 212 equivalent residues and 40% reference coverage.
For all residues but 11 when compared to the super-position based
alignment, it is wrong.
However regions with Insertions and Gaps Can Significantly Vary in Superposition-Based and Statistical Alignments.

Figure 4. Differences in sequence alignment and structure alignment is shown above.
Alignments Based on Superposition
The best feasible foundation for comparative modeling of other family members with unknown structures is provided by sequence alignments based on superposition of the protein family's existing structures.
Deletions / Insertions (indels)
It may be seen via a comparison of sequence alignments and matching superpositions that assumptions about uniform gap penalties do not match up with biological structure.
In contrast to secondary structural components, indels are more frequently seen in loops.
Indels are typically located near the termini of secondary structural components.
Position-specific gap penalties are preferred; gaps are not taken into account by clearly specified secondary structure elements.
many sequence alignment tools provide secondary structure masking as a feature, for instance CLUSTAL.
What a Homology Model is :
A structure-oriented sequence alignment is projected into three dimensions utilizing a related structure (or structures) as a template (s)
Given that 3D structure is far more preserved than sequencing,
The model, which is a 3D map, incorporates knowledge about protein sequences, structural rules governing proteins, and frequently, but not always, evolutionary links.
Building a model
Aligning (many) sequences based on structure should be done first.
Choose an experimental template structure that belongs to the same family as the modeling target and is most similar to it in terms of sequence.
Determine the secondary structure components that are conserved in the template structure (core regions, segments that appear as "blocks" in the structure-based alignment).
The core of the model is composed of conserved areas, including conserved residues.
In core regions, swap out non-conserved residues.
Include backbone and side chain conformations as well as non-conserved areas (loops, twists, indels) on the model's central structure.
By using energy minimization calculations, improve the model's stereochemistry and intramolecular interactions.
Utilize computer visuals, analytical software (PROCHECK), and sequence-structure compatibility programs to evaluate the model.
Rotamer Library: Adding Side Chain Conformations
After the backbone has been modeled, rotamer libraries are used to construct the side chains of non-conserved residues.
As seen in X-ray structures, side chains generally adopt restricted ensembles of distinct low-energy conformations.
X-ray Structures Information
-
geometrical atoms in proteins.
-
Exact conformations of the loop and side chains
-
Specifically arranged catalytic residues
-
patterns of protein structure's hydration
-
proteins' adaptability
-
Upon mutation or ligand binding, conformational changes can occur.
-
Orientations of relative domains
Due to accuracy restrictions and their static nature, models are often unable to properly provide this information.
Observations based on Homology Models
A 3D map to investigate residue surface exposure patterns, residue conservation patterns, and binding site composition.
As an illustration, when choosing residues for site-specific mutagenesis using a model, it would be ideal if the chosen residues mapped to the protein surface, as this preserves the protein's three-dimensional structure while revealing functional locations.
unreliable information from homology model :
-
Bonds, angles, dihedrals,
Side chain conformations,
Local shape, solvent-accessibility values
Information mostly used from homology model :
-
Amino acid locations in core and surface regions,
Spatial relationship of residues in active sites,
Spatial patterns of consen/ed residues,
Surface properties
Information with no guaranteed use cases :
-
Detailed binding site features,
Geometry of catalytic residues,
Functionally important sites,
location of posttranslational modification sites,
suitability as drug target
Numerous Homology Modeling Programs Achieve Similar Levels of Accuracy.
One of the key factors affecting model accuracy is the level of structural conservation of modeling targets and template structures.
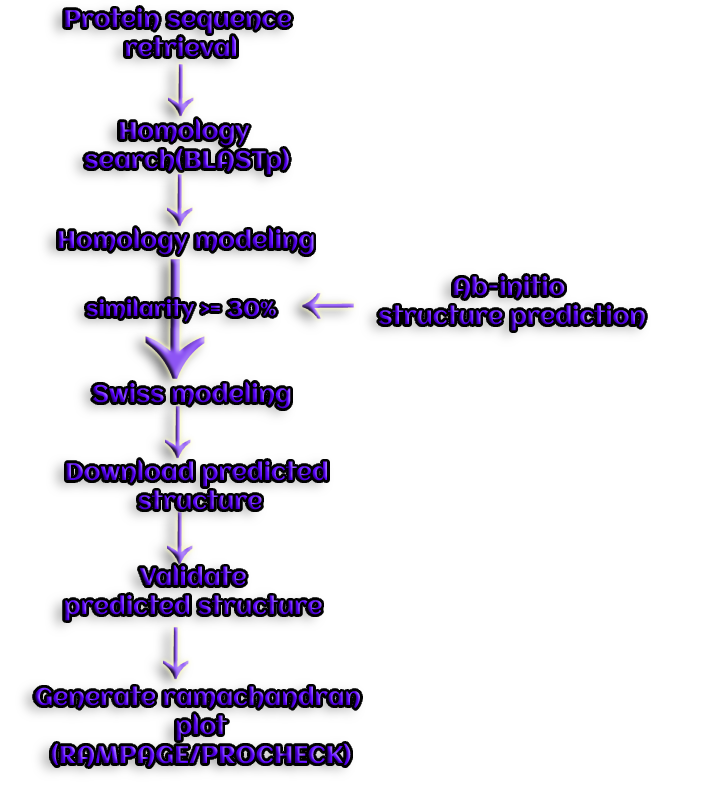
Figure 5. With the aid of homology modeling, a novel structure for a query protein is predicted.
Other Swiss model homology server steps:
-
TAR: Target sequence
-
Search: Sequence database similarity search
-
nr: non-redundant Genbank subset, (with annotated structures)
-
HOM: Homologous sequences
-
TEM: Sequences of homoiogues with known structure
-
Align: Perfonn Multiple Sequence Alignment (MSA)
-
Model: Generate 3D Model
-
EXPDB: Modeling template structure database
-
Complete: Add ligands etc. to model
-
Analyze: interpret and conclude
Model improvement via energy reduction
-
a productive method of "polishing" a protein model.
-
removes structural stresses caused by atomic overlaps and short connections.
-
strengthens or stabilizes hydrogen bonding.
-
transforms protein into a state with low potential energy.
Model improvement using energy efficiency (EM)
-
Even if a model is "folded" wrongly, EM still improves it.
-
Errors resulting from incorrect initial alignment are not fixed by EM.
-
Low potential energy is not always a sign of a good model.
Computational Protein Fold Recognition (Threading)
In some cases, a protein sequence can be accurately assigned a fold by "threading" it onto recognized protein backbone structures.
Similar computations are employed to assess a structure's or model's reliability: is the sequence consistent with the predicted or observed three - dimensional structure? (sequence-structure compatibility)
A method for identifying protein folds called 3D threading involves gradually substituting the sequence of a known protein structure with a query sequence (of unknown structure)
A heuristic measure of protein fold quality is used to evaluate the generated model, and until the best fit is discovered, the process is repeated against all known 3D structures.
The best fit is determined after repeating the method with each known 3D structure.
The fundamental question that necessitates thorough consideration is whether the best fit is "optimal and true" or merely the "best scoring erroneous solution."
Threading
3D structure and sequencing databases; Protein
Data Bank (or non-redundant subset)
Sequence under 25% identity with known structures when queried
Protocol for alignment; dynamic programming
Protocol for evaluation and ranking; Potentials for residue interactions
based on distance
Reasons for threading :
Compared to primary structure, secondary
structure is better conserved.
Compared to secondary structure, tertiary structure is more preserved.
Therefore, rather than using sequence similarity, it is preferable to
use 2D or 3D structural methods to detect remote protein structural
links.
3D threading
Numerous protein sequences correspond to known protein folds.
In light of a database of recognized protein folds:
-
matching each known protein to the test sequence
-
alignment in three-dimensional space (slow)
Score the sequence-structure alignments that
appear.
based on prospective contacts in statistics.
Contact potentials
A protein sequence's capacity to take on a
specific fold is measured by contact potentials.
used mostly for threading (fold recognition)
may also distinguish between structures that are folded appropriately
and those that are not.
Because they are obtained from (non-redundant) collections of PDB structures, contact potentials are empirical.
Deriving a Statistical Contact Potential (Scoring Function)
Set of 3D structures for learning
-
weed out homologous proteins that are redundant.
-
the fold composition is balanced.
-
reference state: randomize sequences on set structures for learning.
Identify the frequency at which each pair of
residues interacts with the learning set.
Remainder interactions, for instance, are counted for 6 distance bins
(relative orientations of interacting residues not taken into account
only distance)
Contact Potential Calculations
Log-odds scores are how statistical potentials are derived.
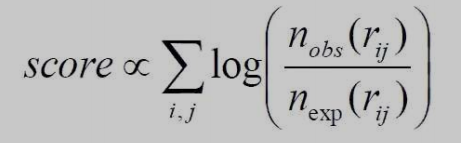
Figure 6. In statistics, the odds ratio's logarithm is known as a log odds. Odds are likelihood ratios that quantify the likelihood that a specific event will occur.
-
totaled across all pairs of interacting residues rij.
-
determined by the log-likelihood of observing each paired interaction in relation to the reference state.
-
interactions between I and j that are frequently binned by ctmance to better distinguish them.
Threading Statistics and Significance
Z—score
-
Standard deviations, number The chosen alignment is not one of the sequence's randomized iterations.
P-value
-
Calculate the distribution of the best random threads and shuffle the sequence.
-
Standard deviations, number The alignment that was selected deviates from how the best threads for each randomized sequence were distributed.
-
The "Z-score of Z-scores"
Database Search Standards of Excellence :
-
Sensitivity
High sensitivity entails searching the database for all potential true matches.
-
Specificity
With great specificity, no false positive matches are found.
Criterion for 3D threading quality
-
extremely sensitive
-
moderate to low specificity
Analysis of the Threading Results
Assume that only one match on a ranked list of high-scoring ones might be accurate.
Expect that any one of these could be true.
The top hits are likely to be false positives.
The selection of true hits frequently involves considering functional similarities as well.
-
Themes in similar sequences
-
Similar binding characteristics or mechanisms
-
Cellular distributions that are similar
Ab-initio Structure prediction
Protein 3D structure prediction without the use
of structural templates or prior information.
Generally speaking, comparative methods are more accurate.
used frequently when fold identification or homology modeling fail.
Comparable to figuring out the "Protein."
There are two main issues:
-
Large conformational space sampling.
-
Global energy minimum determination.
Lattice models and abbreviated chain approaches for the sampling problem.
Threading energies, packing analysis, topology analysis, and molecular mechanics are all involved in the energy problem.
Rosetta
simplified protein structure representations,
rotamer search and fragment insertion (from libraries) are added to ab
initio predictions.
The sampling issue is made simpler by fragment insertion.
Recent advancements made possible by the addition of several homologous
sequences to the query sequences.
The likelihood of achieving a global energy minimum increases with
simultaneous predictions on homologous sequences.
---- Summary ----
As of now you know all basics of Fold Structures.
-
Homology modeling.
-
Threading.
-
Ab-inition structure prediction.
-
etc..
________________________________________________________________________________________________________________________________

________________________________________________________________________________________________________________________________