
Molecular Modeling & Drug Discovery
14. Protein Ligand Docking:
Concept of Docking
Predict protein-ligand complex structures (structure-based drug design)
search databases for fresh active ingredients (structure-based virtual screening)
two significant duties
-
Posing : Predict a ligand's binding conformation and orientation (binding posture) in a protein binding site by posing
-
Scoring : Score tiny compounds based on how likely they are to act as "real" ligands.
Facts that are important:
-
High resolution experimental structure; homology models only if required; reliable 3D structure of protein target.
-
The active site or ligand binding site must be understood.
Docking must produce high form and chemical complementarity of protein-ligand complexes regardless of the methods used.
-
Shape complementarity
-
Chemical complementarity
Modeling the binding site at the molecular level
-
Molecular surface
-
Full atomic (rare)
-
Potential energy grid (similar to the GRID program, grid points store electrostatic and van der Waals potentials)
GRID Approach
Protein is arranged on a grid, various probe atom types are positioned on grid points, and an easy force field is used to calculate interaction energies.
The active site dihydrofolate reductase has potential binding sites that can be mapped using three distinct kinds of probes.
Grid locations with favorable (probe-specific) interaction energies between probes and enzyme atoms combine to create enticing binding sites for small molecule functional groups.
Zone 1 (blue)
is a suitable spot for binding positively charged groups; blue dots
represent the energy function minima from an amino probe.
A desirable location for hydrophobic groups is
Zone 2 (yellow).
Negatively charged groups can bind well in
Zone 3 (red), which is indicated by the red dots that represent
minima from a carboxylate probe.
Docking Methods
Rigid body docking: tiny molecules are docked as rigid conformers to a rigid protein binding site.
Model of "lock-and-key."
Rigid Body Docking
Rigid body docking: tiny molecules have 6 degrees of freedom (3 translational, 3 rotational), but protein coordinates are fixed.
Key and Lock Model
To be potentially active, a tiny molecule conformer (ligand) needs to fit precisely in a protein's binding site (target).
Docking methods
Flexible ligand docking :Protein remains stable, while tiny molecules have the typical 6 Directions of freedoms + internal bond rotation DOF.
It takes longer, but greatly improves the likelihood of accurately predicting real binding conformations.
As mentioned above, flexible protein—ligand docking also allows for the movement of side chain atoms in the binding site.
- A significant structural search space.
- Often approximated by combining various static rotamer combinations, one for flexible ligand docking and the other.
- The main modeling opportunity for local protein conformational changes, but prone to shape mistakes in the binding site
Poses of the ligand within the binding site are
generated by a search algorithm.
How many different ways are there?
-
Three translational and Three rotary
the conformational (torsion angles along single bonds, rotatable bonds)
.png)
Rigid docking
During docking, the conformation of both the
ligand and the receptor is maintained. We just take into account the
six
translational and rotational degrees of freedom.
Through conformational search, a large number of ligand conformations
can be produced without reference to the target. The protein can then be
docked with each ligand shape.
Adaptive ligand docking (most popular approach)
The ligand's conformational space is investigated in search of the most
compatible conformers.
flexible docking of protein and ligand
Both the ligand and the receptor's conformational space are investigated; the side chains in the binding site are adaptable. Additionally, this method requires additional computing.
Flexible Ligand Docking Posing
A variety of ligand conformational sampling methods are used by docking
algorithms:
- Systematic incremental search
- Monte Carlo search
- Molecular Dynamics
- Genetic Algorithms
- Fragment Assembly
- Tabu search
Posing in Rigid and Flexible Docking
Geometry-based Deterministic methods
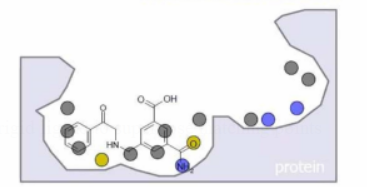
-
Protein: feature points in the cavity
-
Ligand: atoms or feature points
-
Docking: translations +rotations of rigid ligand to superpose matching points
Energy-based Stochastic methods
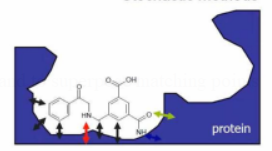
-
Protein : surface atoms / feature points
-
Ligand : atoms or feature points
-
Docking: modification of ligand
Exemplary Geometric Algorithms
DOCK
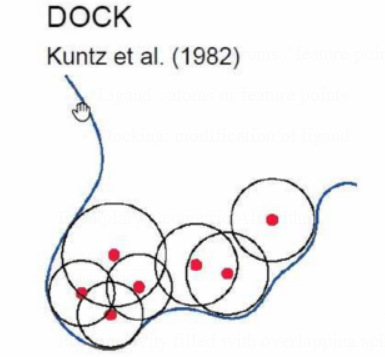
Protein cavity filled with overlapping spheres
(variable radius)
Negative image on binding site
feature points: sphere center
Surflex, MOE

-
Cluster of probes
-
Steric (apolar)
-
polar
-
(NH,CO in surflex, polar in MOE)
Geometric Feature Matching
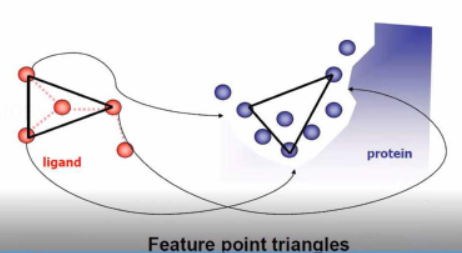
Ligand Flexibility in Geometric Algorithms
Incremental ligand construction (DOCK, Surflex, MOE)
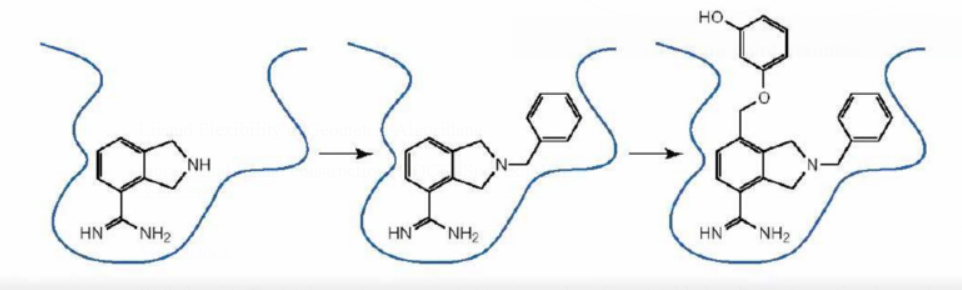
MOE-Dock:
With the aid of a database of recommended torsion values for rotatable bonds, conformational search is coupled to docking.
Exemplary Energy-based Algorithms
Finding stable (low energy) conformations of the protein-ligand combination through optimization of an energy function
Conformer Populations with Iterative generation of conformer populations
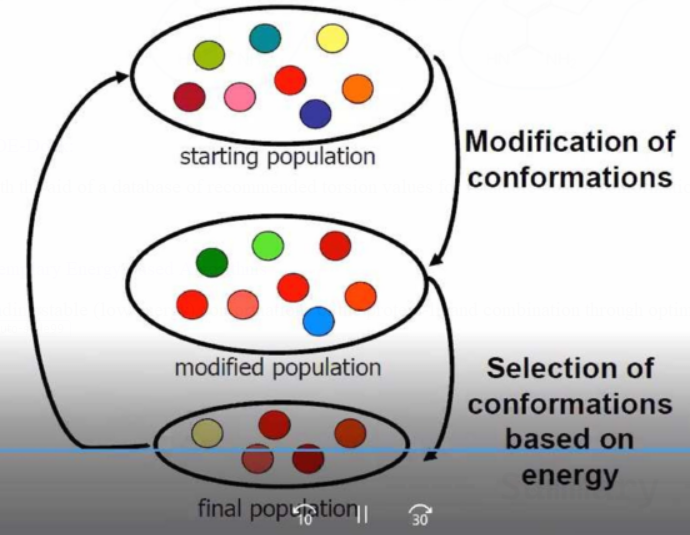
Docking concept
Posing: Binding orientation plus conformation
predicts the binding mode
Evaluation and ranking: Score modeled complexes, separate true hits from
false positives, and place tiny molecules in order of likelihood that
they will act.
Protein—Ligand Interactions
Docking: Non-bonded interactions evaluated
-
Van der Waals interactions
-
Electrostatic interactions
-
Hydrogen bonds
Scoring
Quantum mechanics would provide the most precise
representation of intermolecular interactions
The majority of approaches use molecular mechanics force field (MM
FF)-based methods because of the high processing costs of quantum
mechanics.
Scoring functions
Force fields: usually in vacuum
Knowledge-based: fitting parameters to reproduce interactions in X-ray
structures of complexes
Empirical: fitting parameters to reproduce experimental binding data
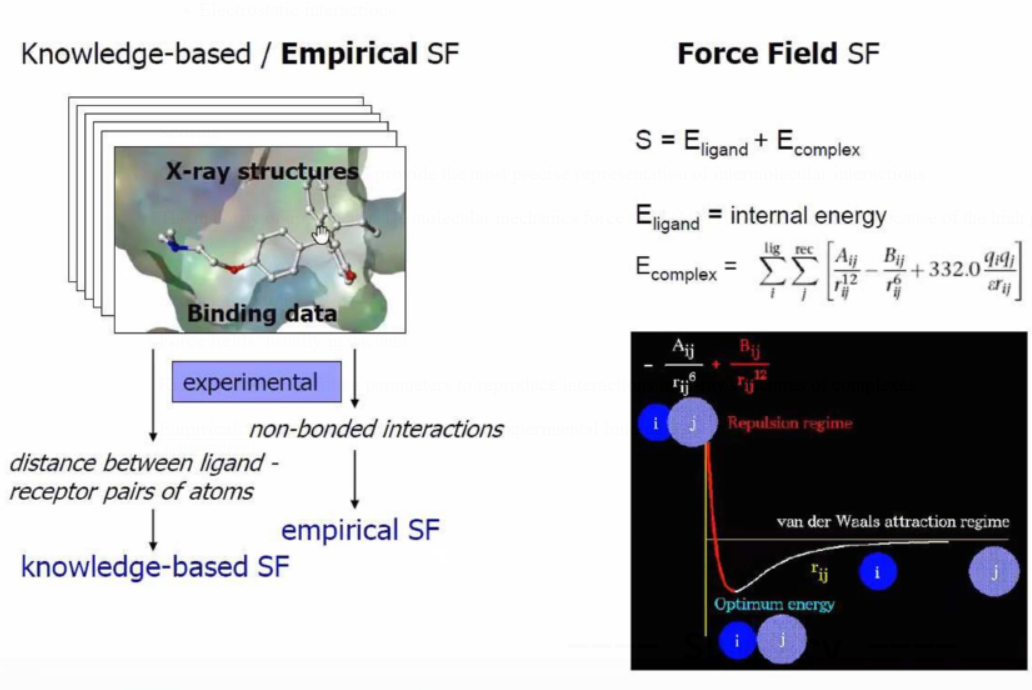
Empirical Scoring Functions
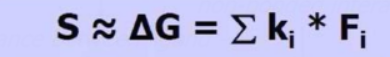
Fi: function which describes protein — ligand interactions computed using geometry predicted by docking
ki : constant fitted using a training set of structures / binding data
MOE Affinity SF
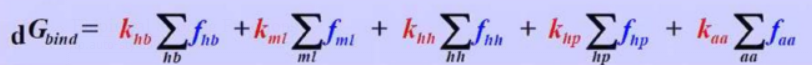
hh :
H-bond donor —
H-bond acceptor
ml : ionic metal - ligand
hh : hydrophobic atom — hydrophobic atoms
hp : hydrophic atom — polar atom
aa : any atom - any atom
k: constant
f: count of atomic contacts


Structure—based Virtual Screening (SBVS)
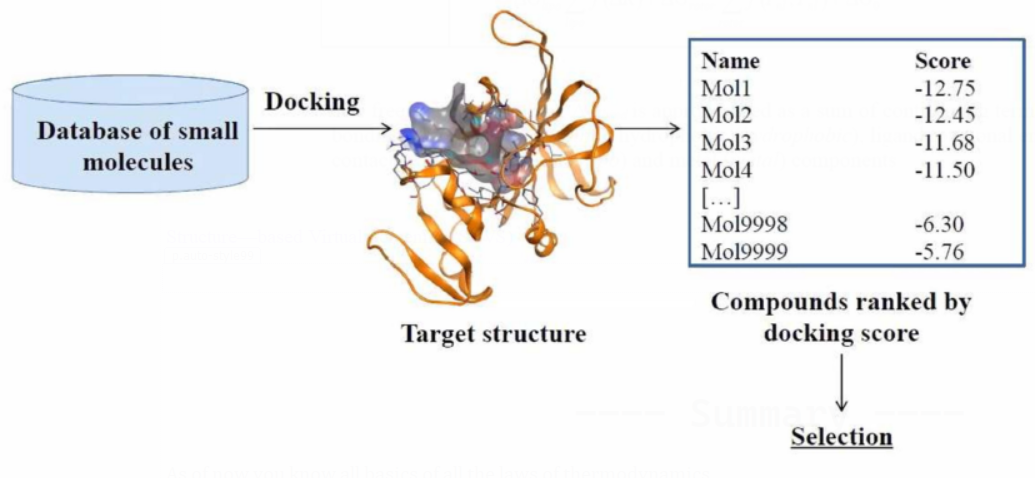
Limitations
Posing:
-
Numerous techniques generate chemically significant postures and replicate known complex structures.
-
However, for the majority of database substances, reasonable postures are constructed.
-
Most substances in the database are false positives.
Scoring:
-
The accuracy of scoring algorithms is typically poor.
-
Experimental poses frequently yield scores that are similar to false positives.
-
A significant issue of docking is the inability to score actual hits systematically better than false—positives.
Scoring Issue
-
Using a force-field based scoring algorithm, the crystal structures of the cofactor ATP and an inhibitor (PIP) attached to a protein kinase were analyzed.
-
The same function was used to dock and score many hundred thousand database substances in the kinase's ATP binding site.
-
A histogram displays the total score distribution. The Gaussian-like distribution's mode corresponds to the scores that are frequently reported for random binding occurrences.
Evaluating Scoring function issues
-
The binding energy is approximated by the FF interaction energies, which are typically calculated in vacuum.
-
The majority of scoring algorithms don't fully account for entropic consequences.
-
Binding-induced conformational changes are not modeled.
-
Beyond training sets, knowledge-based/empirical scoring functions are typically not generalizable.
-
The small number of real positives and the huge number of false positives are not accurately distinguished by the current scoring functions.
Criteria for Database Search Accuracy
Sensitivity: A high sensitivity indicates that
all or almost all of the database's real hits are found.
Specificity: High specificity results in the
search identifying extremely few false positives.
Docking
Medium Sensitivity (often misses true hits)
Low specificity (large number of false positives)
Not posing but scoring is the main issue!
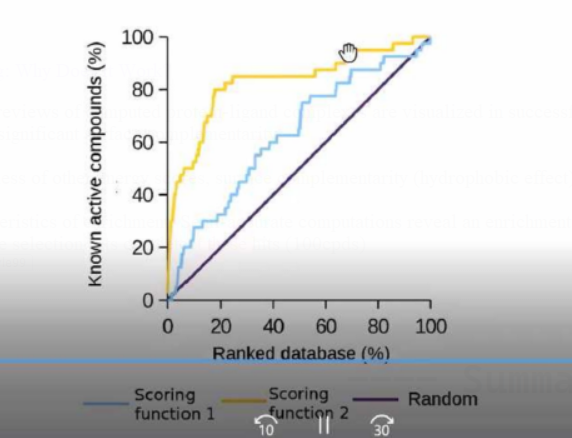
Docking: Why Does It Work?
Expert reviews of computed protein-ligand
complexes are visualized in successful docking experiments (ranking
lists) Protein-ligand complexes often exhibit
significant surface complementarity.
Regardless of other energy scores, surface complementarity (hydrophobic
effect) can be well represented using MM (Lennard-Jones).
Characteristics of enrichment: Semi-accurate computations reveal an
enrichment of hits among better scoring database chemicals. Reasonably
large database selection sets can detect these hits (100cpds)
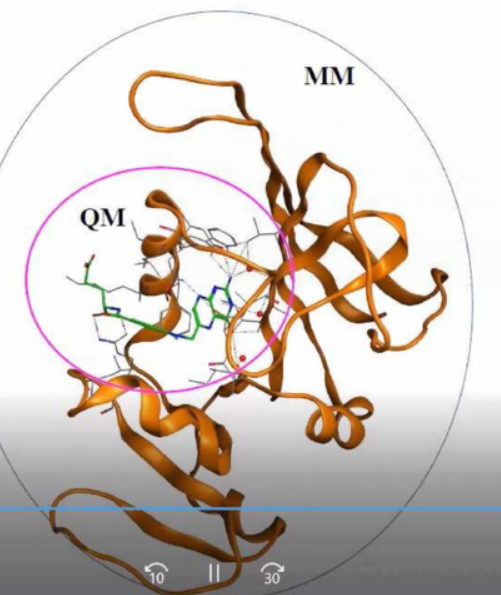
Next Generation Scoring Schemes
QMIMM methods: Treat the active site using QM for accurate energy calculations and the remaining parts of the complex using MM.
Docking in Structure-based Design
Docking considerably helps with compound optimization even though binding energies cannot be precisely calculated:
-
A sequence of analogs can be docked if structures with active chemicals are known.
-
Docked complexes assist in selecting the best analogs for synthesis.
-
Modeled interactions can be used to create new chemical modifications.
-
We obtain hypotheses that are experimentally testable and independent of computed energy ratings.
Active site of cyolooxygenase (COX-2) in complex with :
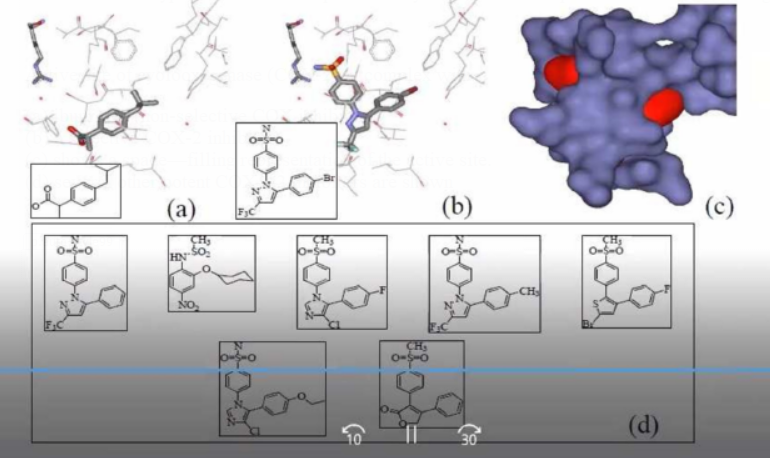
(a) ibuprofen (non-selective COX inhibitor)
(b) a selective COX-2 inhibitor.
(c) shows a space—filling representation of the active site.
(d) several other potent COX—2 inhibitors are
shown
Design of specific inhibitors:
These COX-2 lead compounds have different core structures and functional groups that are compared in the active site using docking to prioritize cpds that match the shape and properties of the active site and suggest chemical modifications.
---- Summary ----
As of now you know all basics of all the laws of thermodynamics.
-
Docking
-
Scoring
-
Emperical scoring functions
-
etc..
________________________________________________________________________________________________________________________________

________________________________________________________________________________________________________________________________