
Molecular Modeling & Drug Discovery
7. Homology modeling:
Homology model
Modeling through homology
involves computational techniques which
can be used to make a prediction for proteins whose structure is
unknown.
The "easiest" (and most well-liked) computational technique for
predicting a protein's structure is called homology modeling (HM).
The objective of HM is to model the structure of the target protein on a
different protein (template) with a sufficient degree of sequence
similarity.
Threading (fold recognition) and ab initio structural prediction are
additional techniques.
Sequence structure data needed for homology modelling :
-
The target protein's sequence.
-
A database of protein structures obtained by experiment (PDB).
Algorithms for sequence alignment can be used to compare a protein's sequence to that of other proteins in the Protein Data Bank (PDB).
If the identity % is in the "safe zone," two protein sequences are likely to adopt the same fold.
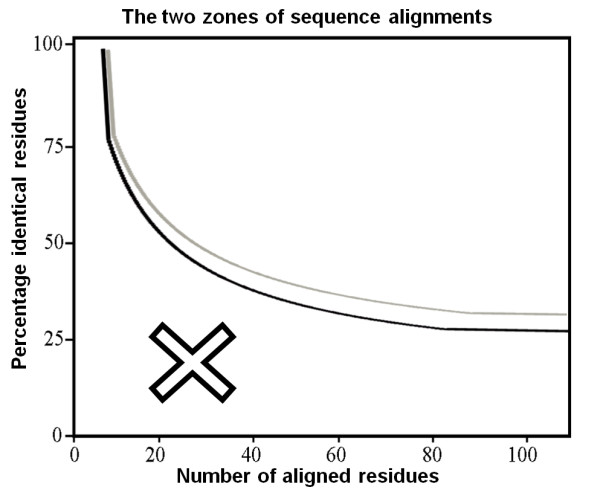
Figure 1. The two sequence alignment identity zones that show whether similar structures are likely to be adopted. If the length and percentage of sequence identity of two aligned sequences occur in the region above the threshold, they have a very high likelihood of having identical folds (black line).
A number of steps go into homology modeling :
-
Template recognition and initial alignment (Initial alignment and recognition of templates)
-
Alignment correction (Adjustment of alignment)
-
Backbone generation
-
Loop modeling (model side chains)
-
Side chain modeling
-
Model optimization (model improvement)
-
Model validation (modeling verification)
1. Initial alignment and template identification
A general guideline for choosing a suitable template is included in safe
homology modeling.
With the aid of sequence alignment tools like BLAST (Altschul et al.,
1990) or FASTA, protein sequences that resemble the query can be found
(Pearson, 1990).
>differences of fasta and blast
The query sequence has been
compared to other proteins' PDB sequences.
The alignments are arranged according to their scores, which are
determined using an alignment matrix.
Typically, the alignment with the greatest score is selected as the
template contender.
Employing various template structures :
-
Different models can be constructed using a variety of templates.
-
One model can be created by combining several templates.
2. Alignment adjustment
For a better alignment, more advanced techniques might be required.
Alignment of several sequences or Multiple Sequence Alignment (MSA)

Figure 2. The alignment of several or more biological sequences (protein or nucleic acid) of comparable length is known as multiple sequence alignment, or MSA. The results allow for the study of the evolutionary links between the sequences and the inference of homology. Programs like ClustalOmega and BLAST may carry out similar tasks.

Score for Blosum 62: -1
The matrix's score remains constant, but this mutation is most likely to occur on the protein's surface, where "Glu" would be exposed to the solvent.
>> insert note BLOSUM PAM matrix
When aligned residues in a multiple sequence alignment are primarily hydrophobic, they are most likely buried in the protein's central region.
Multiple sequence alignments can be used to create scoring matrices (profiles) that are position-specific.
Although the first alignment results in a higher score, the structure suffers as a result.
A structure that can be maintained by small backbone motions results from the second alignment.
3. Backbone Generation
Transform the model sequence
using the coordinates of the template residues that appear in the
alignment.
You can copy the coordinates for 'N', 'Cor', 'C', 'O', and frequently
'CB' as well. To make a first guess, conserved residues can be
replicated entirely.
We give high quality structures priority because even experimental
structures can contain flaws.
Different domains of a single protein can be modeled using a variety of
templates.
4. Loop modeling
There may be gaps in the
alignment of the template and model sequence.
Deletions: (gaps in the model sequence) by leaving residues out of the
template.
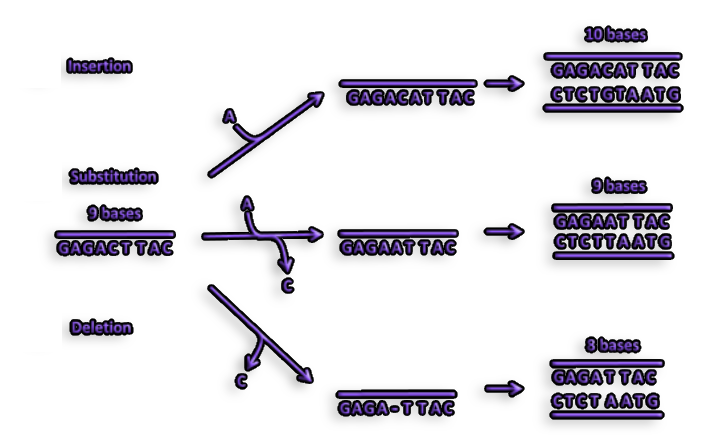
Figure 3. Another kind of mutation known as a frameshift mutation can be caused by the insertion or deletion of one or more nucleotides during replication. A frameshift mutation results in a complete modification of a protein's amino acid sequence.
(Pauses in the template sequence) Insertions: By adding the missing
residues to the continuous backbone, the problem is resolved.
Both scenarios suggest that the backbone's conformation has changed.
These are not likely to occur in typical secondary structural components
(alpha-helices, beta-sheets). although they are more frequent in twists
and loops.
In order to place insertions and deletions inside loops, they are
frequently moved.
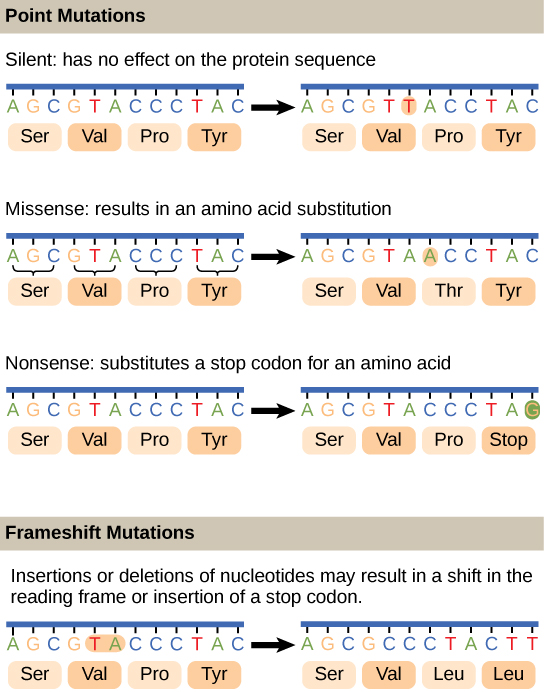
Figure 4. Gene mutations can take place in a gene's nucleotide sequence in two different ways: as point mutations and frameshift mutations. While a frameshift mutation affects one or more nucleotides and modifies the open reading frame of a particular gene, a point mutation only affects a single nucleotide.
Predicting changes in loop conformation is
challenging.
Several elements that may affect loop conformation include:
-
The conformation of surface loops can be impacted by crystal interactions. In the lack of crystallization limitations, a different conformation is anticipated.
-
Depending on the varying steric hindrance of the side chains, modifications to the tiny to bulky residues beneath the loop can change how the loop conforms.
For instance, 'Ala' to 'lle' will probably move the loop farther away. -
Mutations to loop residues: Any response to "Pro" or "Gly" reduces conformational flexibility. A conformational shift of the loop is necessary in order for the new residue to fit into a smaller area of the Ramachandran plot.
Methods for modeling loops:
Knowledge-based: The PDB is searched to find loops with the same end points (a small number of residues) as the loop to be modelled. For the modeling, the detected loop's coordinates are employed.
Energy-based : and includes an ab initio structure prediction. The effectiveness of the loop is assessed using an energy function (force field). Through Monte Carlo simulation or molecular dynamics, the conformation can be improved.
5. Side chain modeling
In proteins that are homologous (identity >
40%), side chain conformations (rotamers) are frequently preserved. The
conserved residues frequently participate in a web of interactions that
control their conformation.
The model simply copies the coordinates of conserved residues from the
template.
To arrange side chains, a library of common rotamers derived from X-ray
structures is frequently employed (knowledge based). An energy function
is then used to assess various conformations.
Position-specific libraries based on backbone
torsion angles and secondary structures (phi,psi) are used to handle
combinatorial explosion.
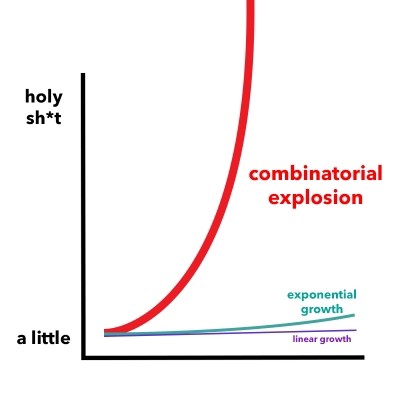
Figure 5. A combinatorial explosion is the rapid increase in a problem's complexity that results from the combinatorics of the issue being influenced by its input, constraints, and boundaries.
The hydrophobic core has a more precise rotamer prediction.
6. Model improvement
A accurately predicted backbone conformation is necessary for an
appropriate prediction of side chains rotamers. However, side chain
packing is essential for the backbone itself.
Iteratively modeling rotamers and backbone structures allows the initial
model to be improved.
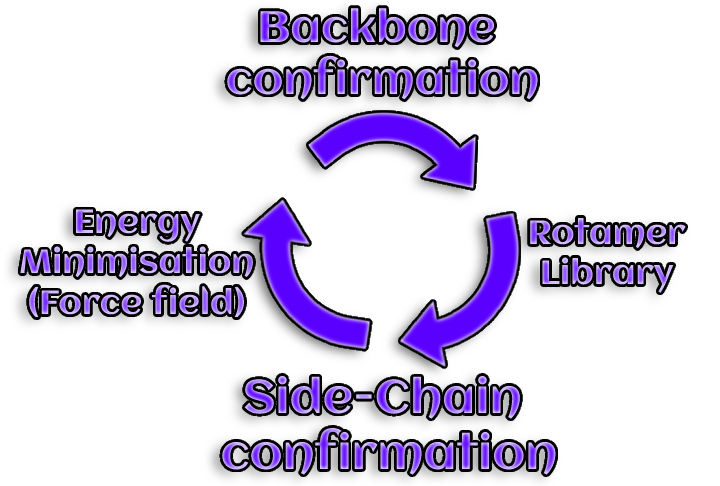
Figure 6. Gene mutations can take pl
Simulations of molecular dynamics can also be utilized to improve the model.
7. Model verification
Errors in a model could be caused by:
percent of the template's and the model's sequences that are identical.
High sequence identity values (> 90%) can result in models that are as accurate as experimentally confirmed structures.
Local model errors are increasingly likely and alignment gets more challenging as identity percentage declines.
Amount of mistakes in the template.
A model's errors can be calculated by calculating normalcy indices with respect to experimental structures. Torsion angles, bond lengths/angles,residue distribution between polar and non-polar, etc..
---- Summary ----
As of now you know all basics of Fold Structures.
-
Homology modeling details
-
7 Steps
-
BLAST,FASTA,PAM,BLOSUM,Mutations
-
etc..
________________________________________________________________________________________________________________________________

________________________________________________________________________________________________________________________________